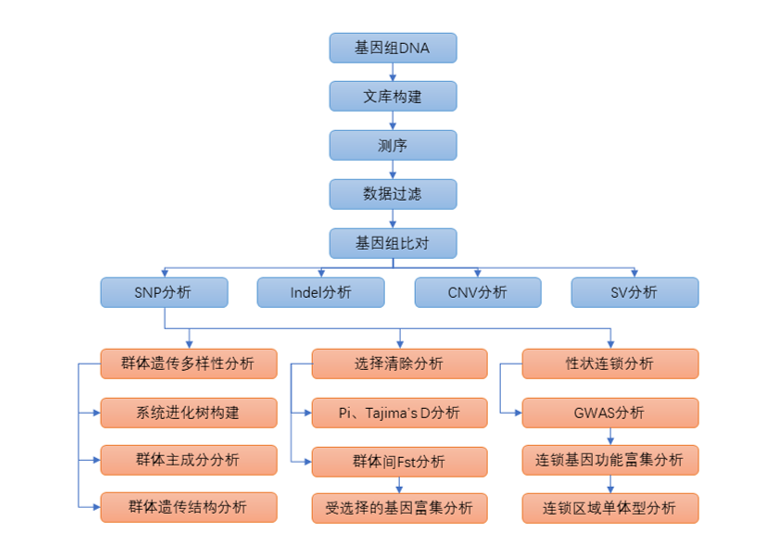
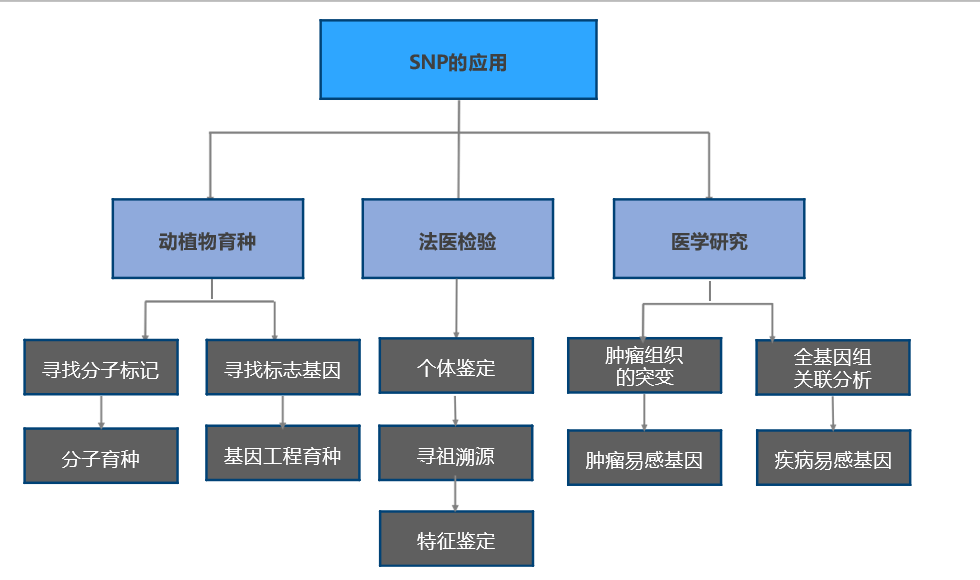
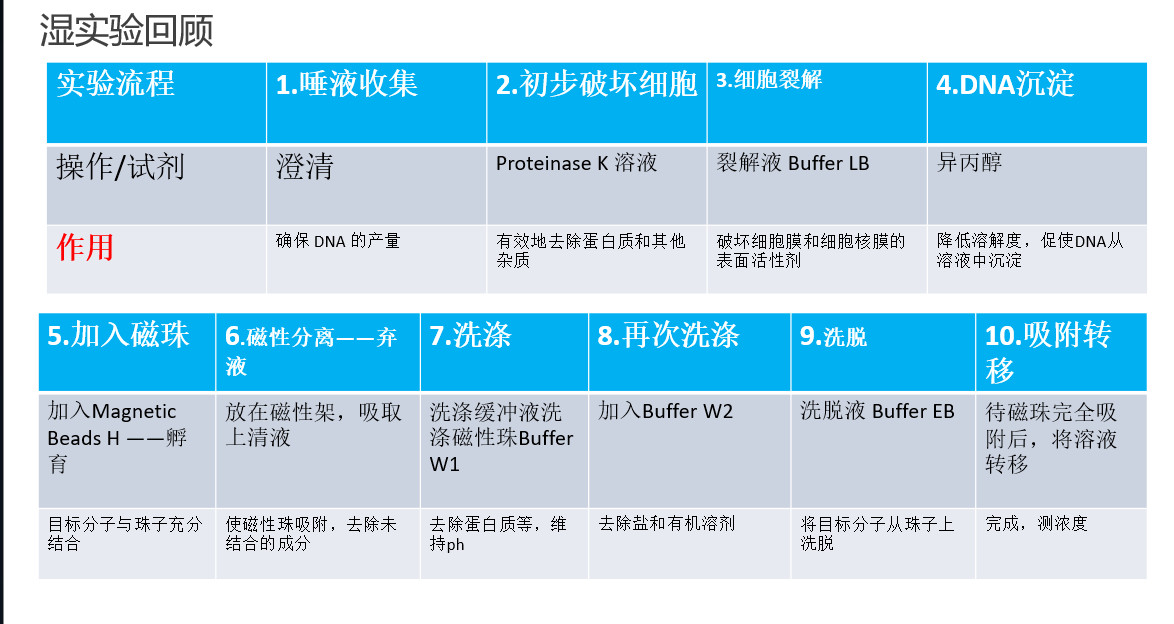
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
326
327
328
329
330
331
332
333
334
335
336
337
338
339
340
341
342
343
344
345
346
347
348
349
350
351
352
353
354
355
356
357
358
359
360
361
362
363
364
365
366
367
368
369
370
371
372
373
374
375
376
377
378
379
380
381
382
383
384
385
386
387
388
389
390
391
392
393
394
395
396
397
398
399
400
401
402
403
404
405
406
407
408
409
410
411
412
413
414
415
416
417
418
419
420
421
422
423
424
425
426
427
428
429
430
431
432
433
434
435
436
437
438
439
440
441
442
443
444
445
446
447
448
449
450
451
452
453
454
455
456
457
458
459
460
461
462
463
464
465
466
467
468
469
470
471
472
473
474
475
476
477
478
479
480
481
482
483
484
485
486
487
488
489
490
491
492
493
494
495
496
497
498
499
500
501
502
| #!/usr/bin/env python3
# -*- coding: utf-8 -*-
"""
The script converts a collection of SNPs in VCF format into a PHYLIP, FASTA,
NEXUS, or binary NEXUS file for phylogenetic analysis. The code is optimized
to process VCF files with sizes >1GB. For small VCF files the algorithm slows
down as the number of taxa increases (but is still fast).
Any ploidy is allowed, but binary NEXUS is produced only for diploid VCFs.
"""
__author__ = "Edgardo M. Ortiz"
__credits__ = "Juan D. Palacio-Mejía"
__version__ = "2.8"
__email__ = "e.ortiz.v@gmail.com"
__date__ = "2021-08-10"
import argparse
import gzip
import random
import sys
from pathlib import Path
# Dictionary of IUPAC ambiguities for nucleotides
# '*' is a deletion in GATK, deletions are ignored in consensus, lowercase consensus is used when an
# 'N' or '*' is part of the genotype. Capitalization is used by some software but ignored by Geneious
# for example
AMBIG = {
"A" :"A", "C" :"C", "G" :"G", "N" :"N", "T" :"T",
"*A" :"a", "*C" :"c", "*G" :"g", "*N" :"n", "*T" :"t",
"AC" :"M", "AG" :"R", "AN" :"a", "AT" :"W", "CG" :"S",
"CN" :"c", "CT" :"Y", "GN" :"g", "GT" :"K", "NT" :"t",
"*AC" :"m", "*AG" :"r", "*AN" :"a", "*AT" :"w", "*CG" :"s",
"*CN" :"c", "*CT" :"y", "*GN" :"g", "*GT" :"k", "*NT" :"t",
"ACG" :"V", "ACN" :"m", "ACT" :"H", "AGN" :"r", "AGT" :"D",
"ANT" :"w", "CGN" :"s", "CGT" :"B", "CNT" :"y", "GNT" :"k",
"*ACG" :"v", "*ACN" :"m", "*ACT" :"h", "*AGN" :"r", "*AGT" :"d",
"*ANT" :"w", "*CGN" :"s", "*CGT" :"b", "*CNT" :"y", "*GNT" :"k",
"ACGN" :"v", "ACGT" :"N", "ACNT" :"h", "AGNT" :"d", "CGNT" :"b",
"*ACGN":"v", "*ACGT":"N", "*ACNT":"h", "*AGNT":"d", "*CGNT":"b",
"*" :"-", "*ACGNT":"N",
}
# Dictionary for translating biallelic SNPs into SNAPP, only for diploid VCF
# 0 is homozygous reference
# 1 is heterozygous
# 2 is homozygous alternative
GEN_BIN = {
"./.":"?",
".|.":"?",
"0/0":"0",
"0|0":"0",
"0/1":"1",
"0|1":"1",
"1/0":"1",
"1|0":"1",
"1/1":"2",
"1|1":"2",
}
def extract_sample_names(vcf_file):
"""
Extract sample names from VCF file
"""
if vcf_file.lower().endswith(".gz"):
opener = gzip.open
else:
opener = open
sample_names = []
with opener(vcf_file, "rt") as vcf:
for line in vcf:
line = line.strip("\n")
if line.startswith("#CHROM"):
record = line.split("\t")
sample_names = [record[i].replace("./", "") for i in range(9, len(record))]
break
return sample_names
def is_anomalous(record, num_samples):
"""
Determine if the number of samples in current record corresponds to number of samples described
in the line '#CHROM'
"""
return bool(len(record) != num_samples + 9)
def is_snp(record):
"""
Determine if current VCF record is a SNP (single nucleotide polymorphism) as opposed to MNP
(multinucleotide polymorphism)
"""
# <NON_REF> must be replaced by the REF in the ALT field for GVCFs from GATK
alt = record[4].replace("<NON_REF>", record[3])
return bool(len(record[3]) == 1 and len(alt) - alt.count(",") == alt.count(",") + 1)
def num_genotypes(record, num_samples):
"""
Get number of genotypes in VCF record, total number of samples - missing genotypes
"""
missing = 0
for i in range(9, num_samples + 9):
if record[i].startswith("."):
missing += 1
return num_samples - missing
def get_matrix_column(record, num_samples, resolve_IUPAC):
"""
Transform a VCF record into a phylogenetic matrix column with nucleotides instead of numbers
"""
nt_dict = {str(0): record[3].replace("-","*").upper(), ".": "N"}
# <NON_REF> must be replaced by the REF in the ALT field for GVCFs from GATK
alt = record[4].replace("-", "*").replace("<NON_REF>", nt_dict["0"])
alt = alt.split(",")
for n in range(len(alt)):
nt_dict[str(n+1)] = alt[n]
column = ""
for i in range(9, num_samples + 9):
geno_num = record[i].split(":")[0].replace("/", "").replace("|", "")
try:
geno_nuc = "".join(sorted(set([nt_dict[j] for j in geno_num])))
except KeyError:
return "malformed"
if resolve_IUPAC is False:
column += AMBIG[geno_nuc]
else:
column += AMBIG[nt_dict[random.choice(geno_num)]]
return column
def get_matrix_column_bin(record, num_samples):
"""
Return an alignment column in NEXUS binary from a VCF record, if genotype is not diploid with at
most two alleles it will return '?' as state
"""
column = ""
for i in range(9, num_samples + 9):
genotype = record[i].split(":")[0]
if genotype in GEN_BIN:
column += GEN_BIN[genotype]
else:
column += "?"
return column
def main():
parser = argparse.ArgumentParser(description=__doc__,
formatter_class=argparse.RawDescriptionHelpFormatter)
parser.add_argument("-i", "--input",
action = "store",
dest = "filename",
required = True,
help = "Name of the input VCF file, can be gzipped")
parser.add_argument("--output-folder",
action = "store",
dest = "folder",
default = "./",
help = "Output folder name, it will be created if it does not exist (same folder as input by "
"default)")
parser.add_argument("--output-prefix",
action = "store",
dest = "prefix",
help = "Prefix for output filenames (same as the input VCF filename without the extension by "
"default)")
parser.add_argument("-m", "--min-samples-locus",
action = "store",
dest = "min_samples_locus",
type = int,
default = 4,
help = "Minimum of samples required to be present at a locus (default=4)")
parser.add_argument("-o", "--outgroup",
action = "store",
dest = "outgroup",
default = "",
help = "Name of the outgroup in the matrix. Sequence will be written as first taxon in the "
"alignment.")
parser.add_argument("-p", "--phylip-disable",
action = "store_true",
dest = "phylipdisable",
help = "A PHYLIP matrix is written by default unless you enable this flag")
parser.add_argument("-f", "--fasta",
action = "store_true",
dest = "fasta",
help = "Write a FASTA matrix (disabled by default)")
parser.add_argument("-n", "--nexus",
action = "store_true",
dest = "nexus",
help = "Write a NEXUS matrix (disabled by default)")
parser.add_argument("-b", "--nexus-binary",
action = "store_true",
dest = "nexusbin",
help = "Write a binary NEXUS matrix for analysis of biallelic SNPs in SNAPP, only diploid "
"genotypes will be processed (disabled by default)")
parser.add_argument("-r", "--resolve-IUPAC",
action = "store_true",
dest = "resolve_IUPAC",
help = "Randomly resolve heterozygous genotypes to avoid IUPAC ambiguities in the matrices "
"(disabled by default)")
parser.add_argument("-w", "--write-used-sites",
action = "store_true",
dest = "write_used",
help = "Save the list of coordinates that passed the filters and were used in the alignments "
"(disabled by default)")
parser.add_argument("-v", "--version",
action = "version",
version = "%(prog)s {version}".format(version=__version__))
args = parser.parse_args()
outgroup = args.outgroup.split(",")[0].split(";")[0]
# Get samples names and number of samples in VCF
if Path(args.filename).exists():
sample_names = extract_sample_names(args.filename)
else:
print("\nInput VCF file not found, please verify the provided path")
sys.exit()
num_samples = len(sample_names)
if num_samples == 0:
print("\nSample names not found in VCF, your file may be corrupt or missing the header.\n")
sys.exit()
print("\nConverting file '{}':\n".format(args.filename))
print("Number of samples in VCF: {:d}".format(num_samples))
# If the 'min_samples_locus' is larger than the actual number of samples in VCF readjust it
args.min_samples_locus = min(num_samples, args.min_samples_locus)
# Output filename will be the same as input file, indicating the minimum of samples specified
if not args.prefix:
parts = Path(args.filename).name.split(".")
args.prefix = []
for p in parts:
if p.lower() == "vcf":
break
else:
args.prefix.append(p)
args.prefix = ".".join(args.prefix)
args.prefix += ".min" + str(args.min_samples_locus)
# Check if outfolder exists, create it if it doesn't
if not Path(args.folder).exists():
Path(args.folder).mkdir(parents=True)
outfile = str(Path(args.folder, args.prefix))
# We need to create an intermediate file to hold the sequence data vertically and then transpose
# it to create the matrices
if args.fasta or args.nexus or not args.phylipdisable:
temporal = open(outfile+".tmp", "w")
# If binary NEXUS is selected also create a separate temporal
if args.nexusbin:
temporalbin = open(outfile+".bin.tmp", "w")
##########################
# PROCESS GENOTYPES IN VCF
if args.write_used:
used_sites = open(outfile+".used_sites.tsv", "w")
used_sites.write("#CHROM\tPOS\tNUM_SAMPLES\n")
if args.filename.lower().endswith(".gz"):
opener = gzip.open
else:
opener = open
with opener(args.filename, "rt") as vcf:
# Initialize line counter
snp_num = 0
snp_accepted = 0
snp_shallow = 0
mnp_num = 0
snp_biallelic = 0
while 1:
# Load large chunks of file into memory
vcf_chunk = vcf.readlines(50000)
if not vcf_chunk:
break
for line in vcf_chunk:
line = line.strip()
if line and not line.startswith("#"): # skip empty and commented lines
# Split line into columns
record = line.split("\t")
# Keep track of number of genotypes processed
snp_num += 1
# Print progress every 500000 lines
if snp_num % 500000 == 0:
print("{:d} genotypes processed.".format(snp_num))
if is_anomalous(record, num_samples):
print("Skipping malformed line:\n{}".format(line))
continue
else:
# Check if the SNP has the minimum number of samples required
num_samples_locus = num_genotypes(record, num_samples)
if num_samples_locus < args.min_samples_locus:
# Keep track of loci rejected due to exceeded missing data
snp_shallow += 1
continue
else:
# Check that neither REF nor ALT contain MNPs
if is_snp(record):
# Add to running sum of accepted SNPs
snp_accepted += 1
# If nucleotide matrices are requested
if args.fasta or args.nexus or not args.phylipdisable:
# Uncomment for debugging
# print(record)
# Transform VCF record into an alignment column
site_tmp = get_matrix_column(record, num_samples,
args.resolve_IUPAC)
# Uncomment for debugging
# print(site_tmp)
# Write entire row of single nucleotide genotypes to temp file
if site_tmp == "malformed":
print("Skipping malformed line:\n{}".format(line))
continue
else:
temporal.write(site_tmp+"\n")
if args.write_used:
used_sites.write(record[0] + "\t"
+ record[1] + "\t"
+ str(num_samples_locus) + "\n")
# Write binary NEXUS for SNAPP if requested
if args.nexusbin:
# Check that the SNP only has two alleles
if len(record[4]) == 1:
# Add to running sum of biallelic SNPs
snp_biallelic += 1
# Translate genotype into 0 for homozygous REF, 1 for
# heterozygous, and 2 for homozygous ALT
binsite_tmp = get_matrix_column_bin(record, num_samples)
# Write entire row to temporary file
temporalbin.write(binsite_tmp+"\n")
else:
# Keep track of loci rejected due to multinucleotide genotypes
mnp_num += 1
# Print useful information about filtering of SNPs
print("Total of genotypes processed: {:d}".format(snp_num))
print("Genotypes excluded because they exceeded the amount "
"of missing data allowed: {:d}".format(snp_shallow))
print("Genotypes that passed missing data filter but were "
"excluded for being MNPs: {:d}".format(mnp_num))
print("SNPs that passed the filters: {:d}".format(snp_accepted))
if args.nexusbin:
print("Biallelic SNPs selected for binary NEXUS: {:d}".format(snp_biallelic))
if args.write_used:
print("Used sites saved to: '" + outfile + ".used_sites.tsv'")
used_sites.close()
print("")
if args.fasta or args.nexus or not args.phylipdisable:
temporal.close()
if args.nexusbin:
temporalbin.close()
#######################
# WRITE OUTPUT MATRICES
if not args.phylipdisable:
output_phy = open(outfile+".phy", "w")
output_phy.write("{:d} {:d}\n".format(len(sample_names), snp_accepted))
if args.fasta:
output_fas = open(outfile+".fasta", "w")
if args.nexus:
output_nex = open(outfile+".nexus", "w")
output_nex.write("#NEXUS\n\nBEGIN DATA;\n\tDIMENSIONS NTAX={:d} NCHAR={:d};\n\tFORMAT "
"DATATYPE=DNA MISSING=N GAP=- ;\nMATRIX\n".format(len(sample_names),
snp_accepted))
if args.nexusbin:
output_nexbin = open(outfile+".bin.nexus", "w")
output_nexbin.write("#NEXUS\n\nBEGIN DATA;\n\tDIMENSIONS NTAX={:d} NCHAR={:d};\n\tFORMAT "
"DATATYPE=SNP MISSING=? GAP=- ;\nMATRIX\n".format(len(sample_names),
snp_biallelic))
# Get length of longest sequence name
len_longest_name = 0
for name in sample_names:
if len(name) > len_longest_name:
len_longest_name = len(name)
# Write outgroup as first sequence in alignment if the name is specified
idx_outgroup = None
if outgroup in sample_names:
idx_outgroup = sample_names.index(outgroup)
if args.fasta or args.nexus or not args.phylipdisable:
with open(outfile+".tmp") as tmp_seq:
seqout = ""
# This is where the transposing happens
for line in tmp_seq:
seqout += line[idx_outgroup]
# Write FASTA line
if args.fasta:
output_fas.write(">"+sample_names[idx_outgroup]+"\n"+seqout+"\n")
# Pad sequences names and write PHYLIP or NEXUS lines
padding = (len_longest_name + 3 - len(sample_names[idx_outgroup])) * " "
if not args.phylipdisable:
output_phy.write(sample_names[idx_outgroup]+padding+seqout+"\n")
if args.nexus:
output_nex.write(sample_names[idx_outgroup]+padding+seqout+"\n")
# Print current progress
print("Outgroup, '{}', added to the matrix(ces).".format(outgroup))
if args.nexusbin:
with open(outfile+".bin.tmp") as bin_tmp_seq:
seqout = ""
# This is where the transposing happens
for line in bin_tmp_seq:
seqout += line[idx_outgroup]
# Write line of binary SNPs to NEXUS
padding = (len_longest_name + 3 - len(sample_names[idx_outgroup])) * " "
output_nexbin.write(sample_names[idx_outgroup]+padding+seqout+"\n")
# Print current progress
print("Outgroup, '{}', added to the binary matrix.".format(outgroup))
# Write sequences of the ingroup
for s in range(0, len(sample_names)):
if s != idx_outgroup:
if args.fasta or args.nexus or not args.phylipdisable:
with open(outfile+".tmp") as tmp_seq:
seqout = ""
# This is where the transposing happens
for line in tmp_seq:
seqout += line[s]
# Write FASTA line
if args.fasta:
output_fas.write(">"+sample_names[s]+"\n"+seqout+"\n")
# Pad sequences names and write PHYLIP or NEXUS lines
padding = (len_longest_name + 3 - len(sample_names[s])) * " "
if not args.phylipdisable:
output_phy.write(sample_names[s]+padding+seqout+"\n")
if args.nexus:
output_nex.write(sample_names[s]+padding+seqout+"\n")
# Print current progress
print("Sample {:d} of {:d}, '{}', added to the nucleotide matrix(ces).".format(
s+1, len(sample_names), sample_names[s]))
if args.nexusbin:
with open(outfile+".bin.tmp") as bin_tmp_seq:
seqout = ""
# This is where the transposing happens
for line in bin_tmp_seq:
seqout += line[s]
# Write line of binary SNPs to NEXUS
padding = (len_longest_name + 3 - len(sample_names[s])) * " "
output_nexbin.write(sample_names[s]+padding+seqout+"\n")
# Print current progress
print("Sample {:d} of {:d}, '{}', added to the binary matrix.".format(
s+1, len(sample_names), sample_names[s]))
print()
if not args.phylipdisable:
print("PHYLIP matrix saved to: " + outfile+".phy")
output_phy.close()
if args.fasta:
print("FASTA matrix saved to: " + outfile+".fasta")
output_fas.close()
if args.nexus:
output_nex.write(";\nEND;\n")
print("NEXUS matrix saved to: " + outfile+".nex")
output_nex.close()
if args.nexusbin:
output_nexbin.write(";\nEND;\n")
print("BINARY NEXUS matrix saved to: " + outfile+".bin.nex")
output_nexbin.close()
if args.fasta or args.nexus or not args.phylipdisable:
Path(outfile+".tmp").unlink()
if args.nexusbin:
Path(outfile+".bin.tmp").unlink()
print( "\nDone!\n")
if __name__ == "__main__":
main()
|